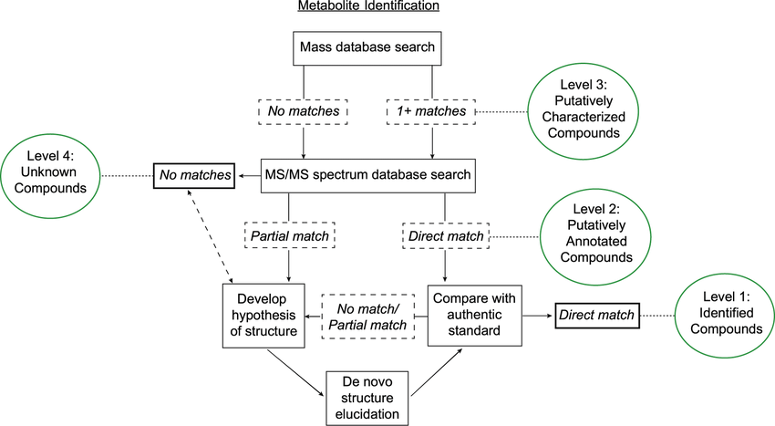
Compound identification in metabolomics
Starting with a m/z
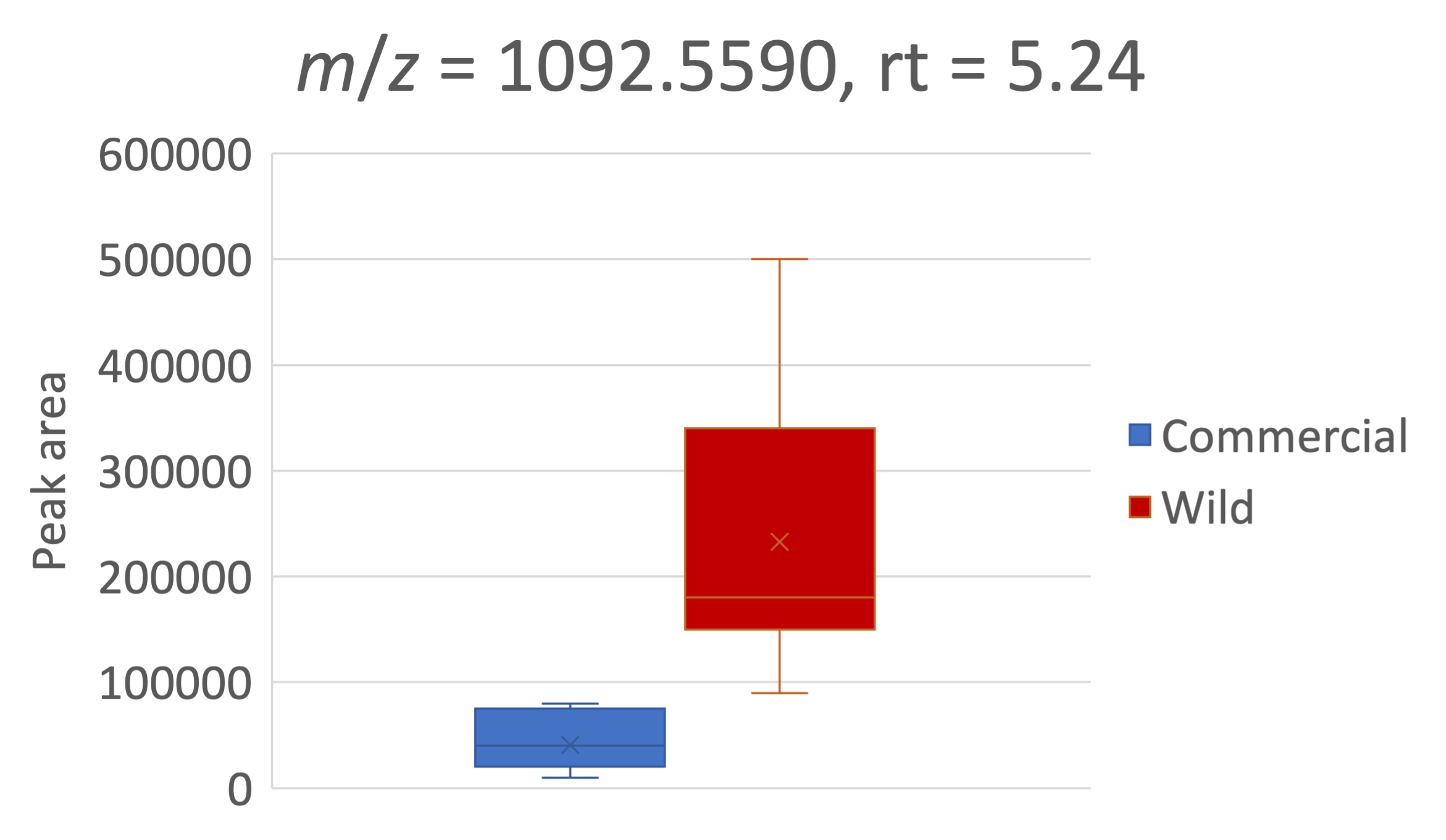
- A feature of interest differentially present in commercial vs. wild tomatoes. We want to know what this is.
- UHPLC-QTOF-MS, ESI+, reversed phase C18, Methanol extract
- A useful adduct calculator
Search in MS databases - HMDB
Search > LC-MS Search
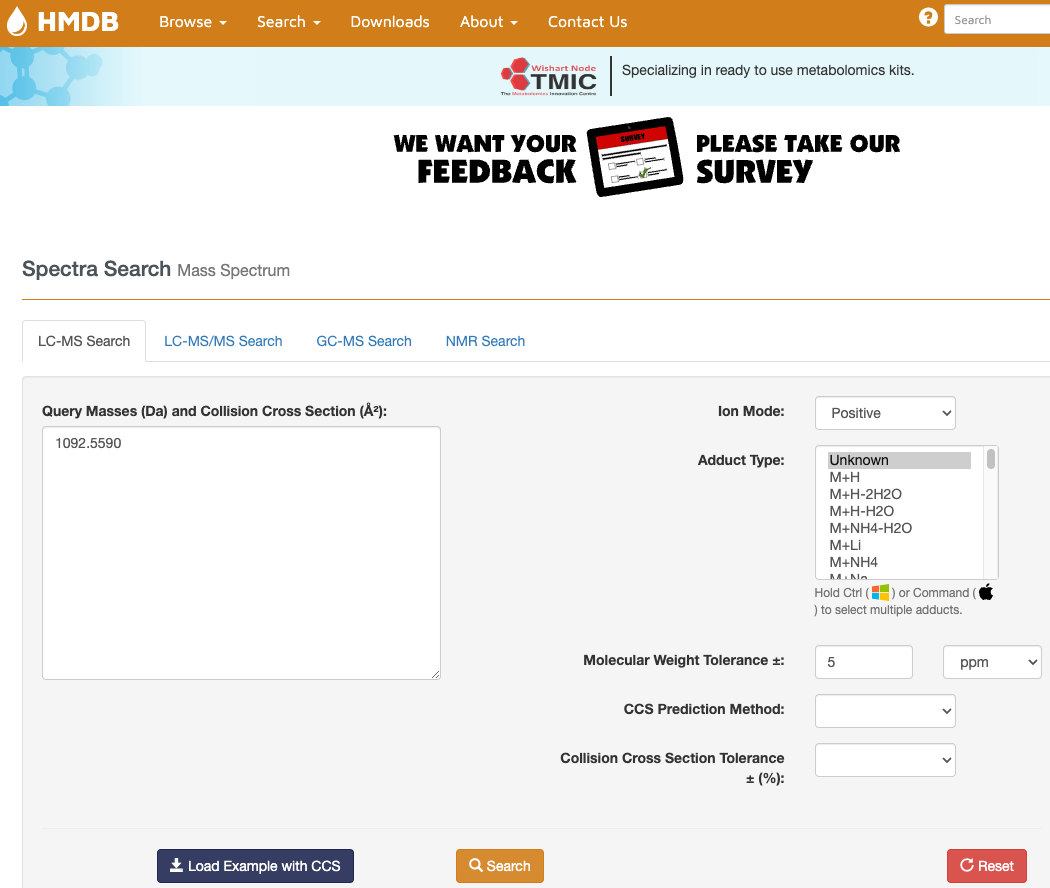
- Enter your mass and mode
- Indicate adduct type (can put unknown if you don’t know)
- Select a mass error (5 ppm is good for a QTOF)
- Search
Access or collection MS/MS fragmentation data

MS/MS spectra of 1092.5590 at 65eV in +ESI
Download structure
http://www.hmdb.ca/spectra/ms_ms/59530
Download an .sdf file of your structure
Import into ChemSketch
Import your .sdf file into ChemSketch
Set ChemSketch to provide you structural information
- Select which parameters you want printed:
Tools>Calculate>Select properties to calculate> Select “Molecular Formula”, “Monoisotopic Mass”, “[M+H]+” - Have ChemSketch calculate those parameters:
Tools>Calculate>Selected properties>Copy to editor.
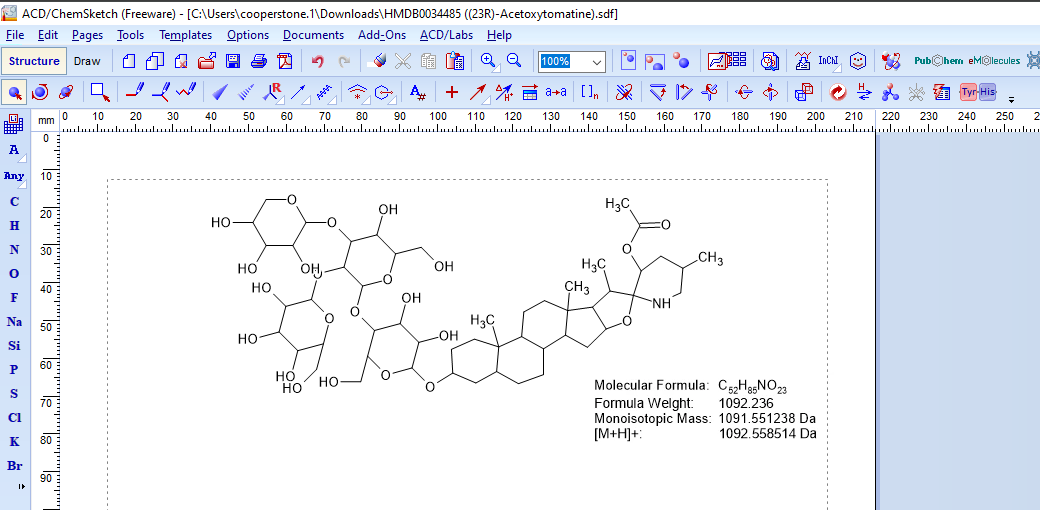
Breaking bonds to rationalize fragments
- Use the eraser (delete) to break bonds (learn more here re: using ChemSketch)
- Select a bond and it will be deleted
- The details will stay with the larger fragment. Highlight the smaller piece to recalculate parameters.
- Account for any rearrangement or additions/subtractions
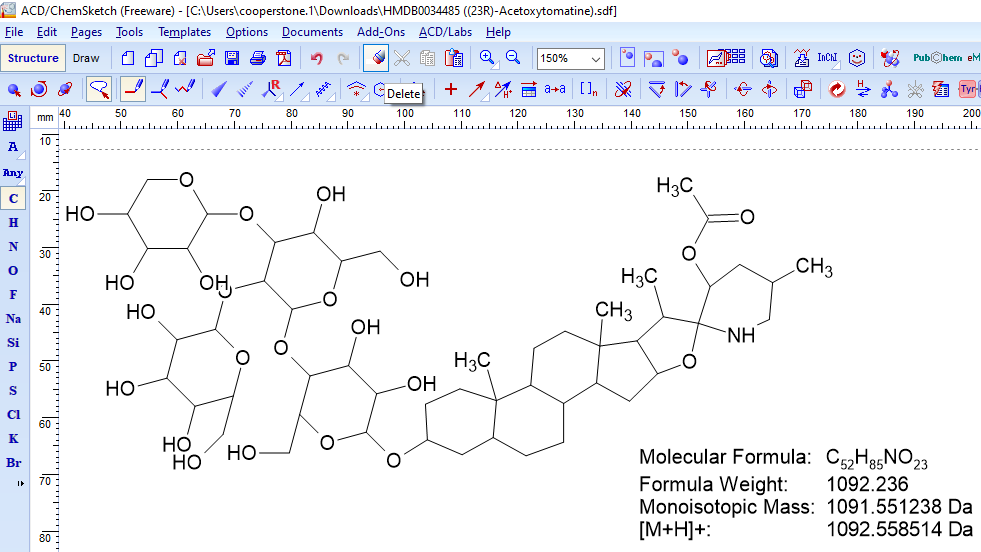
Breaking bonds to rationalize fragments
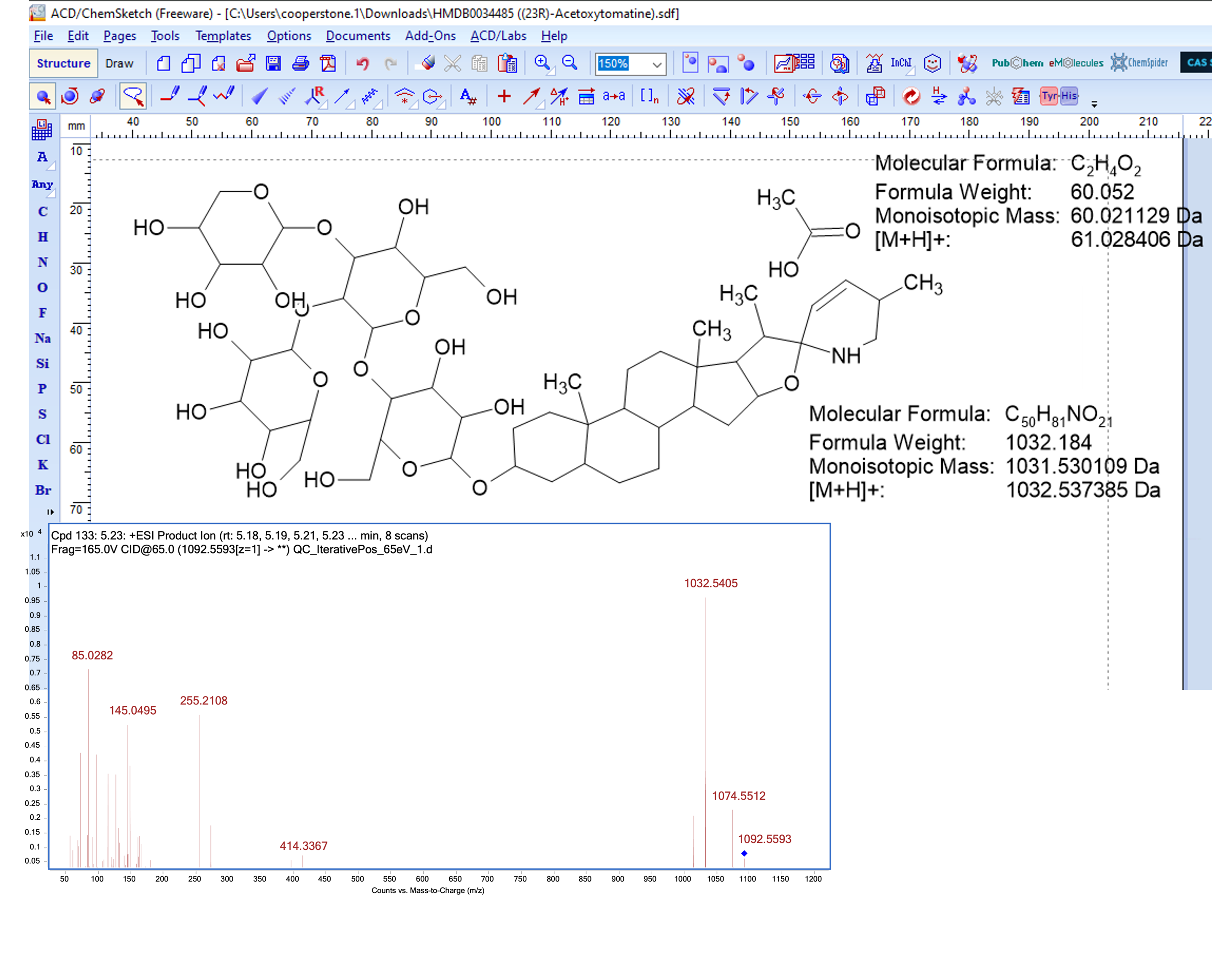
Cleaving at the acetoxy group
Breaking bonds to rationalize fragments
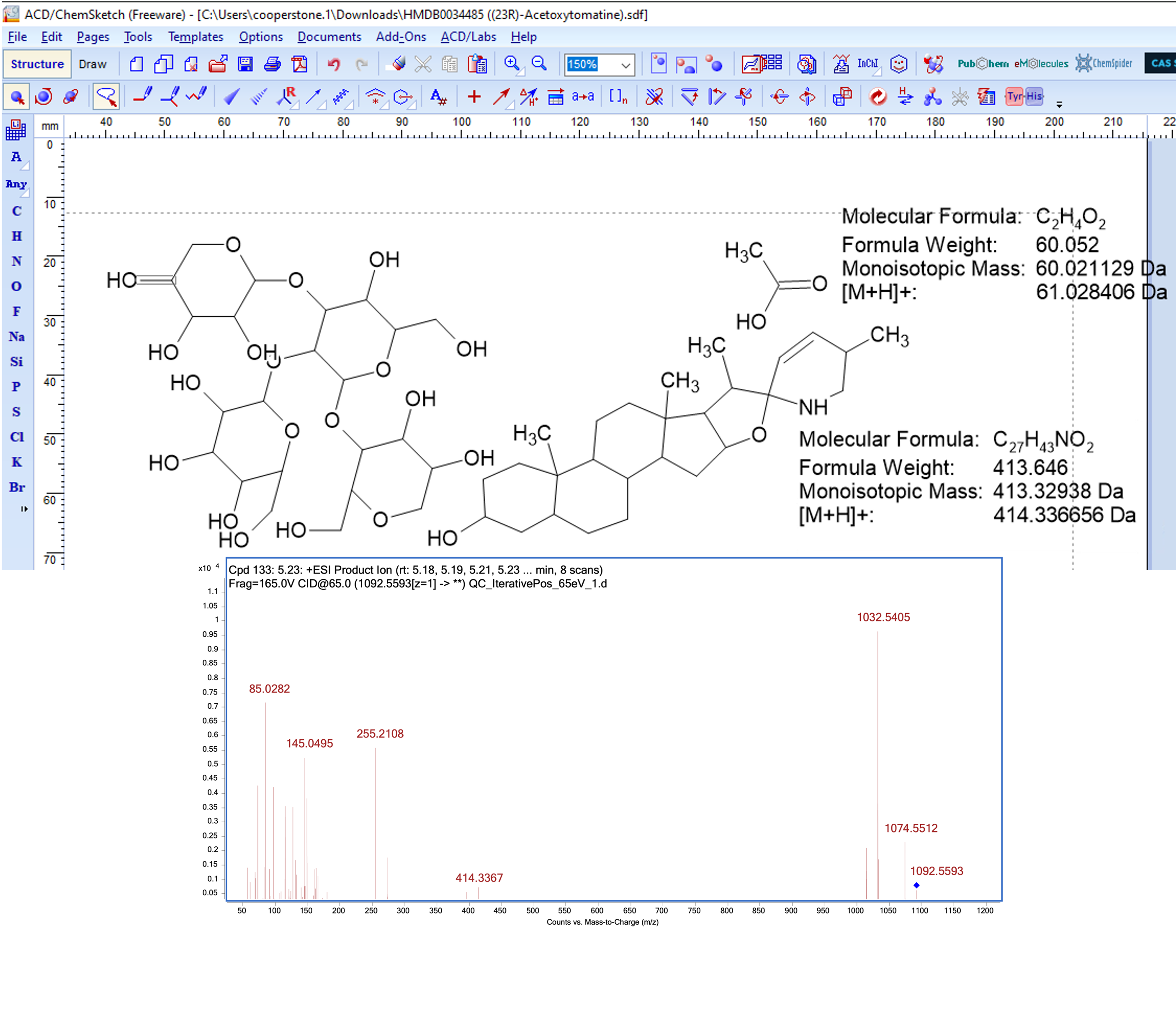
Cleaving at the acetoxy group
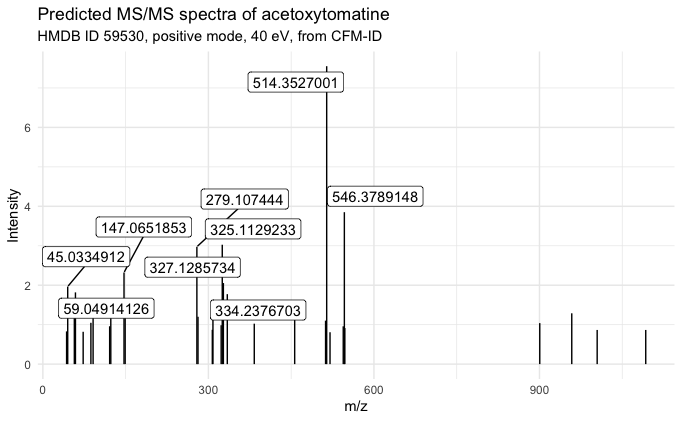
MS/MS predicted spectra of acetoxytomatine
http://www.hmdb.ca/spectra/ms_ms/59530
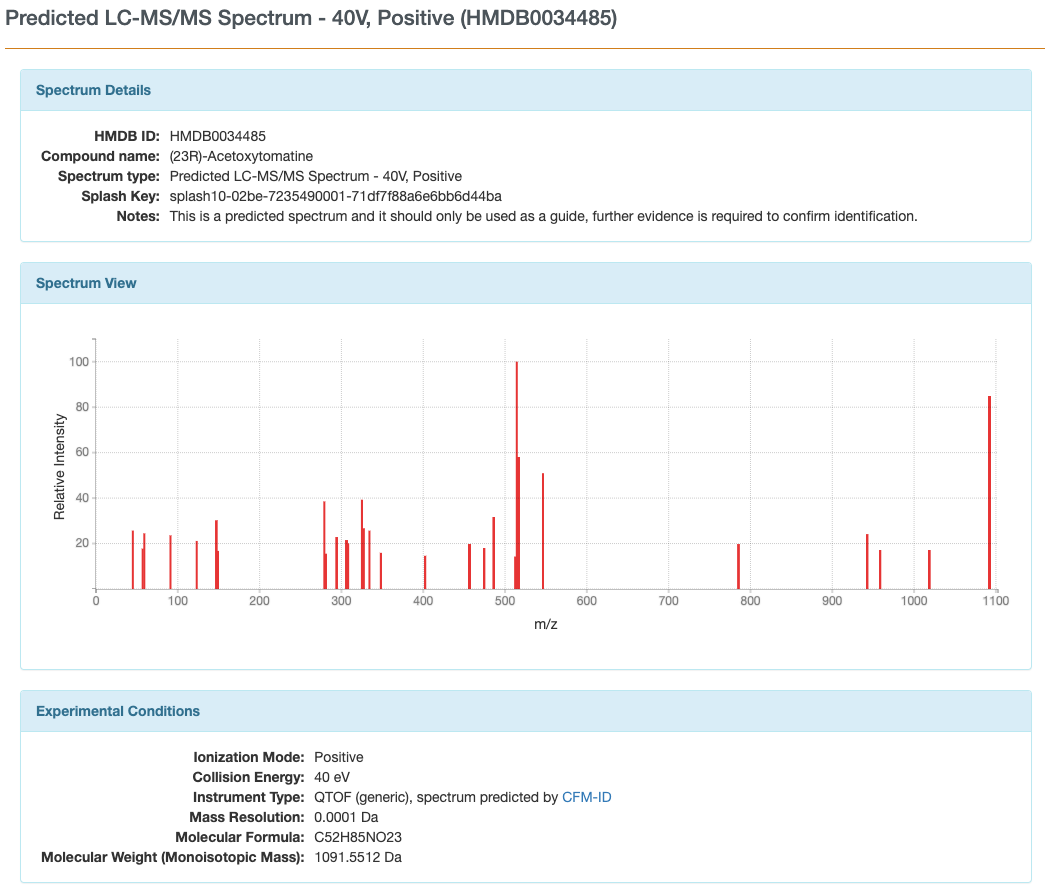
Predicted spectra for (23R)-acetoxytomatine
Predicted vs. actual
LC-MS metabolite ID workflow
Do you need to have an ID to quantify?